Untargeted Metabolomics Pre-processing#
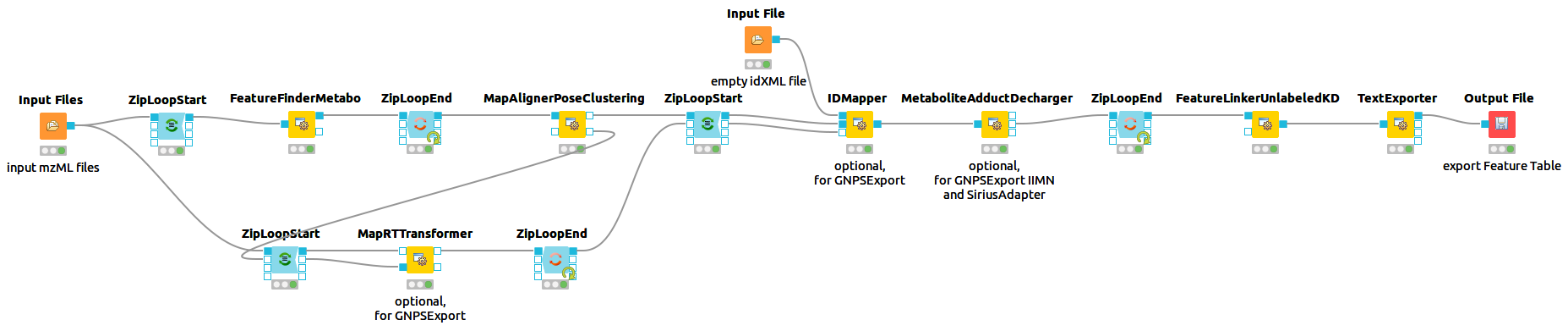
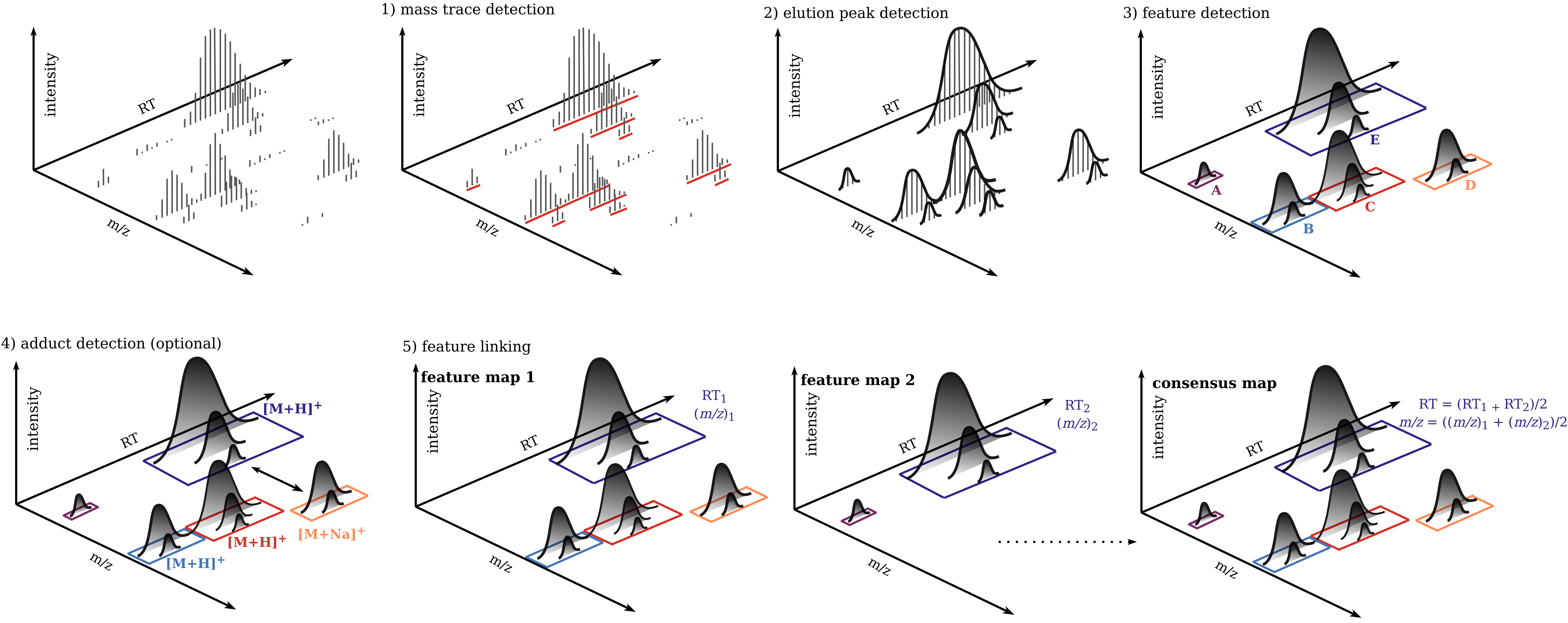
The universal workflow for untargeted metabolomics always consists of feature detection in the individual MS sample files and their linkage to consensus features with common m/z and retention time values. In addition, there are optional steps such as adduct detection and annotation of features with associated MS2 spectra. This workflow prepares all the file necessary to do formula and structural annotations via SiriusAdapter. Furthermore it prepares all required files to run GNPSExport, which generates all files necessary to directly run GNPS Feature Based Molecular Networking (FBMN) and Ion Identity Molecular Networking (IIMN).
If you want to use the example data, download the files sample1.mzML and sample2.mzML.
For each mzML file do mass trace, elution peak and feature detection.
FeatureFinderMetabo -in sample1.mzML -out sample1.featureXML -algorithm:common:noise_threshold_int 10000 -algorithm:mtd:mass_error_ppm 10 -algorithm:ffm:remove_single_traces true
FeatureFinderMetabo -in sample2.mzML -out sample2.featureXML -algorithm:common:noise_threshold_int 10000 -algorithm:mtd:mass_error_ppm 10 -algorithm:ffm:remove_single_traces true
Align feature retention times based on the feature map with the highest number of features (reference map).
MapAlignerPoseClustering -in sample1.featureXML sample2.featureXML -out aligned_sample1.featureXML aligned_sample2.featureXML -trafo_out sample1.trafoXML sample2.trafoXML -algorithm:pairfinder:distance_MZ:max_difference 10.0 -algorithm:pairfinder:distance_MZ:unit ppm
Align mzML files aligment based on FeatureMap alignment (optional, only for GNPS).
MapRTTransformer -in sample1.mzML -out aligned_sample1.mzML -trafo_in sample1.trafoXML
MapRTTransformer -in sample2.mzML -out aligned_sample2.mzML -trafo_in sample2.trafoXML
Map MS2 spectra to features as PeptideIdentification objects (optional, only for GNPS). Requires an empty idXML file.
IDMapper -id empty.idXML -in aligned_sample1.featureXML -spectra:in aligned_sample1.mzML -out IDmapped_sample1.featureXML
IDMapper -id empty.idXML -in aligned_sample2.featureXML -spectra:in aligned_sample2.mzML -out IDmapped_sample2.featureXML
Detect adducts (optional, only for SIRIUS and GNPS Ion Identity Molecular Networking).
MetaboliteAdductDecharger -in IDmapped_sample1.featureXML -out_fm adducts_sample1.featureXML -algorithm:MetaboliteFeatureDeconvolution:potential_adducts "H:+:0.6" "Na:+:0.1" "NH4:+:0.1" "H-1O-1:+:0.1" "H-3O-2:+:0.1"
MetaboliteAdductDecharger -in IDmapped_sample2.featureXML -out_fm adducts_sample2.featureXML -algorithm:MetaboliteFeatureDeconvolution:potential_adducts "H:+:0.6" "Na:+:0.1" "NH4:+:0.1" "H-1O-1:+:0.1" "H-3O-2:+:0.1"
Link features in a ConsensusMap.
FeatureLinkerUnlabeledKD -in adducts_sample1.featureXML adducts_sample2.featureXML -out Preprocessed.consensusXML -algorithm:link:rt_tol 30.0 -algorithm:link:mz_tol 10.0
Export table of metabolic features as tsv file including meta values (e.g. best consensus adduct ion).
TextExporter -in Preprocessed.consensusXML -out Features.tsv -consensus:add_metavalues
You can recreate this workflow in KNIME. Download the KNIME workflow here. The workflow should look like this: